Research Reveals Excessive NR Can Disrupt Mouse Heart Mitochondria
A study in eLife shows that nicotinamide riboside (NR) might have disadvantageous effects on the mitochondria of heart cells, demonstrating the importance of NAD+ precursor choice.
Highlights
· High levels of mitochondrial damage can cause a drop in NAD+ levels, which ultimately drives a breakdown of function and communication in cardiac mitochondria.
· Treatment with the NAD+ precursor NR does not get the desired effect of re-establishing protein activity dependent on NAD+.
· These data suggest that the use of NR in cardiac disorders should be examined more closely, in particular at higher dosages.
Heart failure is a pressing worldwide public-health problem with millions of patients having worsening heart function. Although heart failure is complex, dysfunction of mitochondria — our cells’ batteries — seems to be an important target for therapy to improve cardiac function directly.
In a study published in eLife, a research team based out of Oslo University Hospital in Norway provide evidence of a disease-causing mechanism that connects mitochondrial damage to heart dysfunction via reduced levels of NAD+, an abundant and vital co-enzyme critical to mitochondrial function. Using mice, Lauritzen and colleagues demonstrate that high levels of cardiomyocyte mitochondrial damage cause a reduction in NAD+ levels.
In addition, high dosages of the NAD+ precursor nicotinamide riboside (NR) worsen the mitochondrial damage in the heart. Consequently, the Norwegian researchers conclude that high doses of NR should be used with caution, especially when cardiomyopathic symptoms are caused by mitochondrial dysfunction.
“These data also suggest that the use of NR in rescuing these cardiac events should be reevaluated, in particular at higher dosages,” conclude Lauritzen and colleagues.
The link between mitochondrial damage and NAD+
Mitochondria contain their own genome, called mitochondrial DNA (mtDNA). Though it is uncertain how the instability of mtDNA affects mitochondrial function during the development of heart dysfunction, mtDNA must remain relatively damage‐free.
DNA damage is predominantly recognized by enzymes called poly(ADP-ribose) polymerases (PARPs) that initiate repair mechanisms. PARP activity depends on nicotinamide adenine dinucleotide (NAD)+ as a substrate and localizes to the mitochondria and the nucleus. High PARP activity has been speculated to drain NAD+ reserves.
Another family of enzymes called sirtuins, which work by modifying proteins through a process called deacetylation, is central in the cellular defense against DNA damage and oxidative stress. Sirtuins are shown to promote longevity and can mitigate many diseases related to aging and cardiovascular disease, such as heart failure. Consequently, depletion of the NAD+ pool would cause cellular harm due to loss of sirtuin activity and dysregulation of several protective pathways.
SIRT3 is particularly relevant to heart failure since it resides in mitochondria where it regulates many mitochondrial proteins. Loss of SIRT3 activity due to suboptimal NAD+ levels leads to deregulation of essential and varied molecular processes, including antioxidant systems, mtDNA repair, and mitochondrial dynamics.
Can NAD+ precursors help treat heart failure?
Agents that could maintain NAD+ levels could therefore be an attractive therapeutic approach in disorders where increased PARP activity and decreased SIRT activity drive impaired mitochondrial function and increased mtDNA damage, and ultimately cellular and tissue failure. A recent study showed that supplementation with the NAD+ precursor nicotinamide mononucleotide (NMN) could partially normalize NAD+ levels and restore sirtuin activity and consequently mitochondrial function in mice with heart failure.
Also, studies using nicotinamide riboside (NR) to boost NAD+ levels have recently shown beneficial effects for several physiological functions, including cardiac function in mice with dilated cardiomyopathy. However, these compounds’ therapeutic benefits and safety are far from clear, and if and how NR supplementation improves cardiac function needs to be further elucidated.
Unstable NAD+ metabolism leads to impaired heart mitochondrial function.
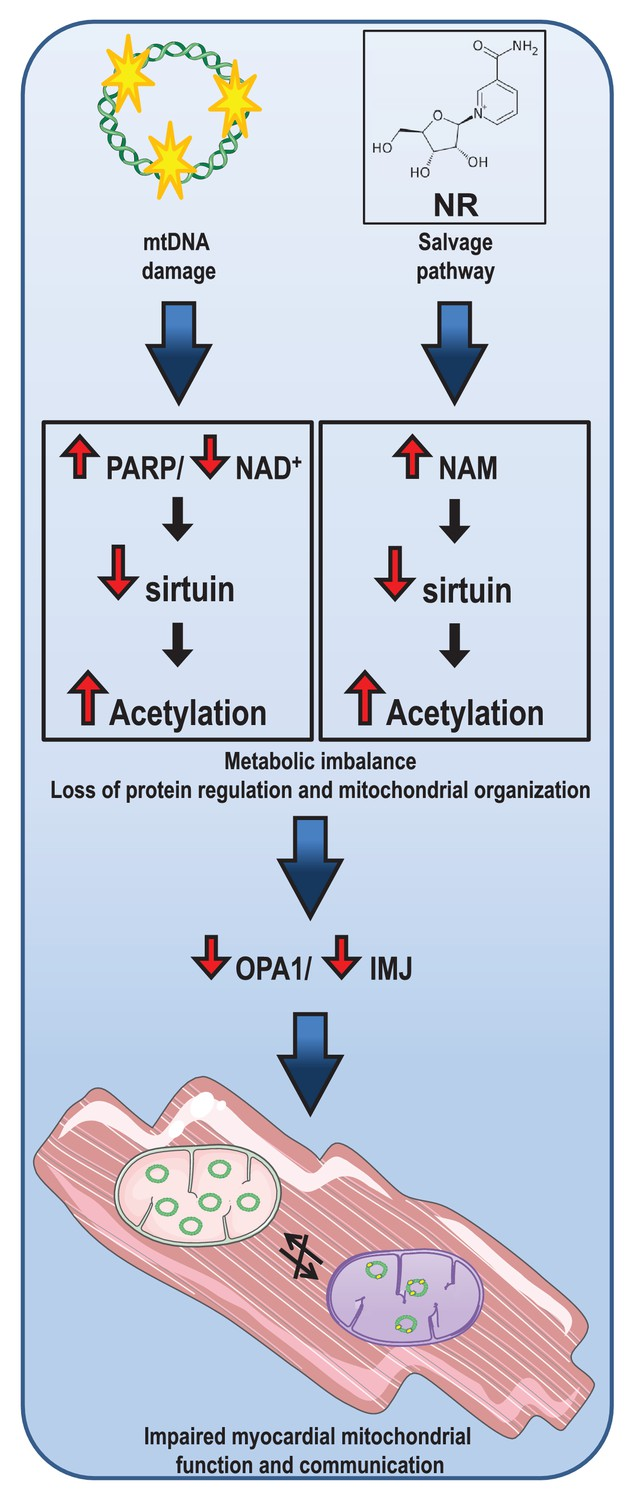
Lauritzen and colleagues show that increased PARP activity in response to increased mtDNA damage depleted NAD+ levels in mice with a mutated mitochondrial component (mutUNG1) that drives heart failure. These interactions resulted in decreased mitochondrial deacetylation, most likely due to impaired SIRT3 activity, which promoted further mitochondrial dysfunction, potentially representing a disease-causing loop in the progression of cardiac remodeling in these mice.
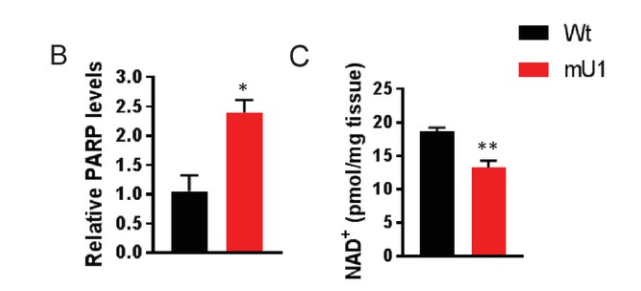
Lauritzen and colleagues found that NR supplementation to increase NAD+ levels failed to improve the cardiac mitochondrial characteristics in the mutUNG1 mice. While NR has been suggested to restore NAD+ levels and enhance mitochondrial function and SIRT3 activity, the Norwegian researchers show that NR, particularly at high doses, had the opposite effects in cardiac tissue. Moreover, detailed studies of the mitochondria show that NR promoted the disorganization of mitochondrial structures.
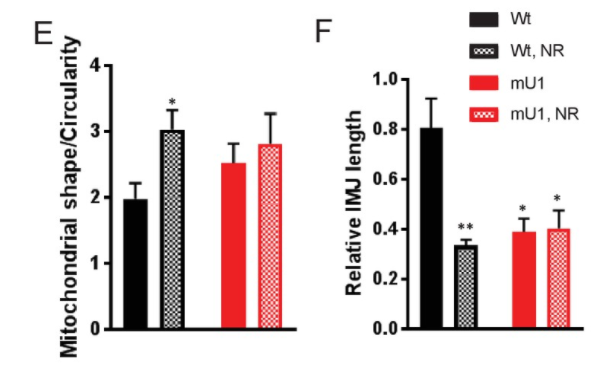
A higher dosage of NR even worsened the cardiac deacetylation status in these mice. Importantly, NR caused a marked increase in NAM in both cardiac and liver tissue, and this compound has been shown to inhibit sirtuin activity. It is, therefore, likely that NAM-mediated inhibition of sirtuin activity in heart tissue during NR supplementation results in impaired rather than improved cardiac protein deacetylation and function.
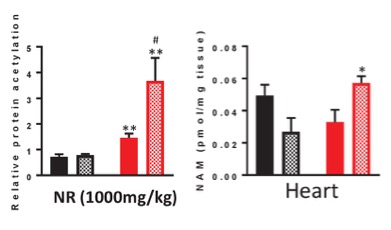
NR has recently become a popular tool for boosting NAD+ levels and has been reported to have anti-aging properties and suggested as a treatment for patients suffering from brain disorders including Alzheimer’s disease. However, in this study, NR does not seem to have overall positive effects. On the contrary, NR supplementation induced a failing of cristae structures to align between neighboring mitochondria, and the IMJ sites seemed to be lost.
Lauritzen and colleagues conclude, “Consequently, high doses of NR should be used with caution, especially when cardiomyopathic symptoms are caused by mitochondrial dysfunction and instability of mtDNA.”
What about using other NAD+ precursors like NMN to treat heart failure?
The question then becomes whether application of any NAD+ precursor would benefit heart failure patients, in light of the evidence that NAD+ levels get depleted in this scenario. A study in 2017 showed that short-term administration of nicotinamide mononucleotide (NMN) preserves cardiac mitochondrial homeostasis and prevents heart failure. Although this study used a different mouse model, these mice also presented with protein hyperacetylation and reduced Sirt3 and NAD+ levels. The authors demonstrated that short-term administration of NMN successfully protected the mutant mice from pressure overload-induced heart failure and that NMN preserved mitochondrial ultrastructure, reduced ROS, and prevented cell death in the heart.
Collectively, these results provide convincing evidence that hyperacetylation of mitochondrial proteins is critical in the pathogenesis of cardiac disease and that administration of NMN may serve as a promising therapy. So, it’s not necessarily the case that all NAD+ precursors may negatively affect mitochondrial heart failure patients; it’s just about picking the right one. You may want to steer clear of NR regarding the heart’s health and, instead, go ahead with NMN.
 Comments
Comments