Study Shows NMN May Treat Rare Transmissible Neurodegenerative Disorder
Increasing NAD+ levels with nicotinamide mononucleotide (NMN) improves the defective clearance of damaged mitochondria caused by prion diseases, protecting these neurons from cell death.
Highlights
- Mitophagy – the clearance of damaged mitochondria – is impaired in a mouse cell model of a rare neurodegenerative condition known as a prion disease.
- Supplementation with NMN or urolithin A (UA) in neuronal cells with prion disease significantly revives mitophagy.
- However, only NMN and not UA could alleviate prion-induced mitochondrial damage and dysfunction as well as neuronal cell death.
Prion diseases, which caught the public eye due to the 2003 “mad cow disease” outbreak in the UK, are a group of chronic, fatal, neurodegenerative diseases that can infect humans and animals. As with many other neurodegenerative diseases, especially those that are age-related, prion diseases – or transmissible spongiform encephalopathies (TSEs) – involve misfolded proteins, like Alzheimer’s and Parkinson’s disease, are often affected by a persistent accumulation of damaged mitochondria in neurons.
Researchers from China Agricultural University in Beijing, China, show that two NAD+ boosting compounds protect prion disease neurons from death. Published in Cell Death & Disease, Li and colleagues show that NMN and urolithin A improve the pool of healthy mitochondria in prion disease neurons by inducing mitophagy – the process for clearing damaged mitochondria. These findings suggest that mitophagy-stimulating compounds like these compounds that boost NAD+ could be therapeutic interventions for prion diseases.
Mangled Mitochondria May Perpetuate Prion Disease
The name “prions” refers to aberrant, disease-causing agents that are transmissible and can cause improper folding of particular normal cellular proteins known as prion proteins, which are prevalent in the brain. Prion diseases, such as Creutzfeldt-Jakob disease, may occur spontaneously, be inherited, or be transmitted by contact with infected tissue, such as during a transplant or from eating contaminated meat.
While the roles of these normal prion proteins are currently being researched, it’s known that the aberrant folding of prion proteins causes brain damage and the disease’s typical signs and symptoms. Prion diseases have been shown to cause personality changes, anxiety, depression, and memory loss, usually within a few months, and many people lapse into coma. Prion illnesses generally advance quickly and are invariably deadly, but how this rapid neurodegeneration occurs remains a bit of a mystery.
A persistent accumulation of damaged mitochondria in neurons has been associated with aging and neurodegenerative diseases, including prion diseases. Mitophagy, one of the intracellular mitochondrial quality control pathways, can selectively remove damaged mitochondria, which may be associated with the accumulation of damaged mitochondria in prion diseases. Many studies have reported the importance of PINK1-parkin dependent mitophagy pathways in neurons: PINK1-parkin forms a signal transduction pathway that labels damaged mitochondria for elimination.
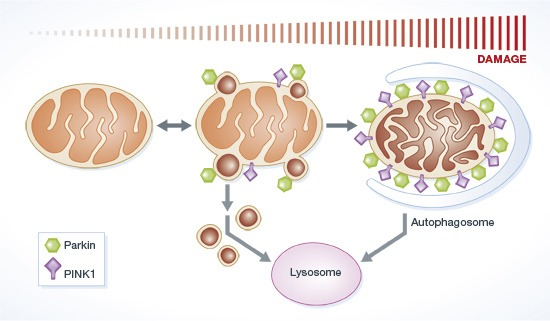
(McLelland et al., 2014 | The EMBO Journal) Parkin and PINK1 function in regulating mitochondrial quality control. In response to mitochondrial stress, Parkin induces the PINK1-dependent formation of mitochondrial vesicles that target cell structures called lysosomes that envelop cargo for degradation. This process is called mitophagy.
NAD+ Boosters Improve Mitophagy in Prion Disease Neuronal Cells
Since mitochondrial damage is an early indicator of neuronal damage, discovering the connections between mitophagy deficiency and mitochondrial damage is very important for the early prevention and treatment of prion diseases. In this study, Li and colleagues investigated damaged mitochondrial accumulation in a prion disease cell model. For the model, the Beijing-based research team infected mouse neuronal cells with a toxic protein fragment that affects prions, called prion protein 106-126 (PrP106-126).
This treatment caused extensive cell death, decreasing the viability by 50-60%. Additionally, the mitochondria were visibly fragmented, indicative of severe mitochondrial damage, and the PINK1-Parkin-mediated mitophagy was defunct and hardly active. But when treated with NMN, the prion-modeling neuronal cells were protected from death. In contrast, UA could not significantly alleviate PrP106-126-induced cell death.
When Li and colleagues genetically manipulated the prion modeling neuronal cells to produce excess Pink1 and Parkin, the mitophagy deficiency was alleviated. Similarly, supplementation with two mitophagy-inducing agents, nicotinamide mononucleotide (NMN) or urolithin A (UA), significantly stimulated PINK1-Parkin-mediated mitophagy. However, compared with NMN, UA could not alleviate prion-induced mitochondrial fragmentation and dysfunction.
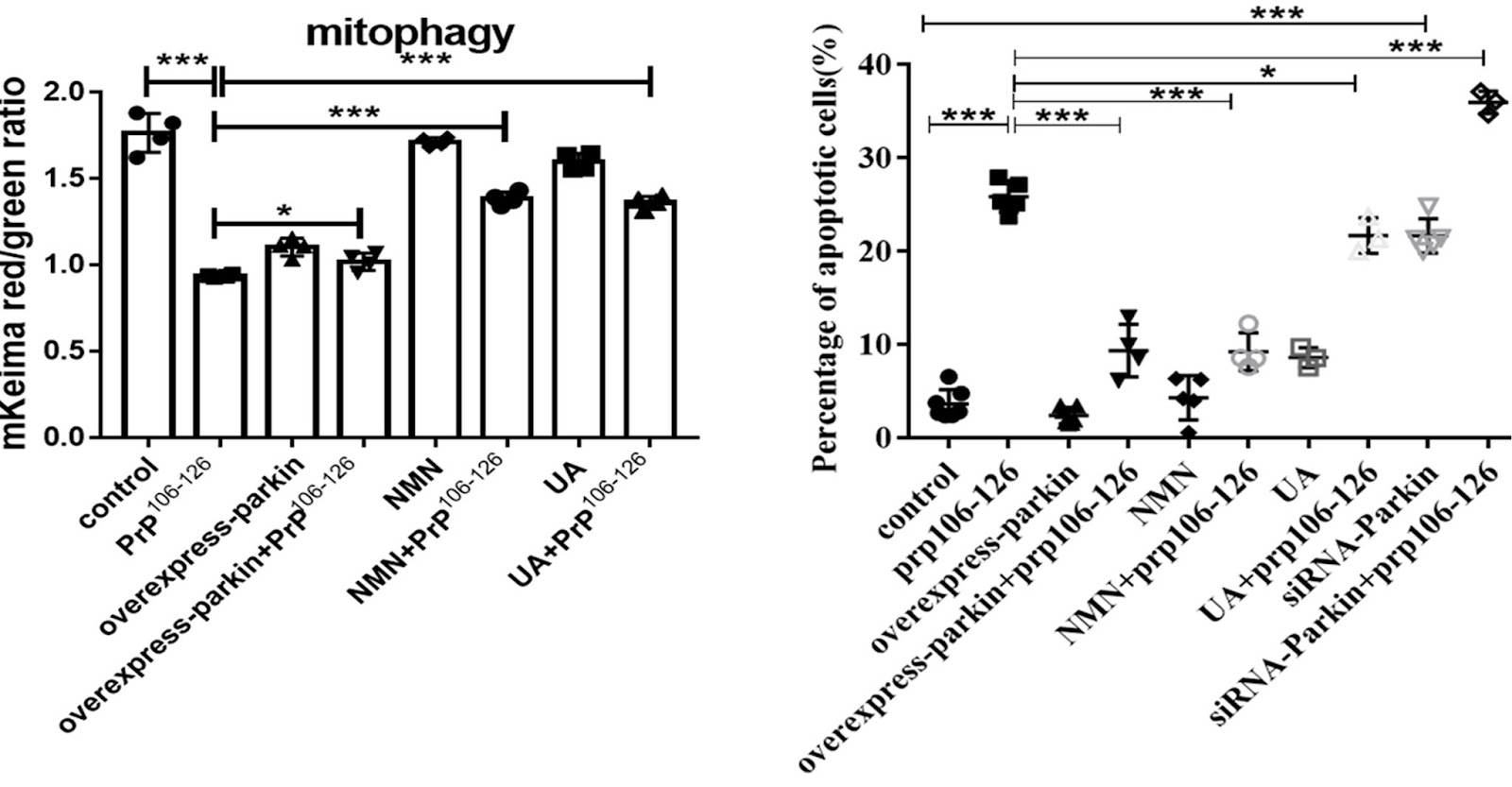
(Li et al., 2022 | Cell Death & Disease) Activation of mitophagy with NMN, but not urolithin A, attenuates cell death in neurons modeling prion disease. Li and colleagues tested the effects of NMN and urolithin A (UA) on mitochondrial fragmentation and cell viability in neuronal cells treated with or without being infected by a prion disease-causing agent (PrP106-126).
Why Does NMN but Not Urolithin Protect Prion Disease Neurons From Death?
Li and colleagues show that cells affected by prion disease have severe mitochondrial fragmentation and dysfunction. The mitophagy activator NMN activates PINK1-Parkin-mediated mitophagy in the prion disease cell model and restores mitochondrial morphology and function, thereby alleviating neuronal cell death induced by PrP106-126. The therapeutic effect of NMN has also been reported in other diseases, such as premature aging-related ataxia telangiectasia and hypertension-related stroke.
On the other hand, while UA activates mitophagy, it does not alleviate prion-affected cells from damaged mitochondrial fragmentation. The resulting cells are left with few functional mitochondria for UA to function. That’s why the Beijing-based researchers think that UA supplementation wasn’t able to alleviate neuronal death caused by prion disease. UA cannot alleviate mitochondrial fragmentation and dysfunction caused by PrP106-126, eventually leading to cell death and decreased vitality, an interesting phenomenon.
The different mechanisms of NMN and UA in activating mitophagy require further study, which may help us find precise targets for the treatment of prion diseases like Creutzfeldt-Jakob disease. Since defects in this mitophagy pathway have been reported in other diseases, like Alzheimer’s and Parkinson’s disease as well as cardiomyopathies, mitophagy activators may have broad therapeutic potential. Different mitophagy activators may work better to treat specific diseases, so there is a need to screen for the mitophagy activators that promote cell health and viability in each specific disease context.
 Comments
Comments